
H. Hnátová, L. Hornofová, T. Adla, J. Veselka
Souhrn
Představujeme kazuistiku 36leté pacientky s akutní myokarditidou v rámci eozinofilní granulomatózy s polyangiitidou. Pacientka s anamnézou asthmabronchiale a četnýchalergií byla vyšetřena na urgentním příjmu našeho zařízení s klinickými projevyakutní myokarditidy a významnou leukocytózou s eozinofilií v krevním obraze. Vevzorku získaném endomyokardiální biopsií z pravé komory byla popsánanekrotizující eozinofilní myokarditida.
Eozinofilní granulomatóza s polyangiitidou jejednou z etiologií způsobujících hypereozinofilní syndrom. Jedná se o vzácnésystémové autoimunitní onemocnění a postižení srdce může být jednou z jehosoučástí.
Podezření na toto onemocnění by mělo být vzneseno u pacientů sesymptomy a příznaky myokarditidy a/nebo perikarditidy nebo s poruchami srdečníhorytmu, které jsou spojeny s eozinofilií v krevním obraze, s anamnézou asthmabronchiale, alergické rinitidy, abnormalitami paranazálních sinů a neuropatiemi.Léčba eozinofilní granulomatózy s polyangiitidou je založena na imunosupresi,zejména kortikosteroidy, ale zváženy mohou být i další látky, zejména azathioprin.
© 2020, ČKS.
Klíčová slova:
Eozinofilie
Eozinofilní granulomatóza s polyangiitidou
Eozinofilní myokarditida
Abstract
We present a case– clinical presentation, diagnosis, management and follow up of a 36-year-old woman with acute myocarditis as a manifestation of eosinophilic granulomatosis with polyangiitis (EGPA). The patient with a history of asthma and allergies presented with clinical signs of acute myocarditis and with peripheral eosinophilia in blood, an endomyocardial biopsy was then performed with histological proof of necrotizing eosinophilic myocarditis.
EGPA is one of the various causes of hypereosinophilic syndromes. It is a rare multisystemic autoimmune disorder and cardiac involvement can represent one of its components.
The suspicion should be raised in patients with symptoms and signs of myocarditis and/or pericarditis or heart rhythm abnormalities with eosinophilia in blood count, with a history of asthma, allergic rhinitis, abnormalities in the paranasal sinuses and neuropathy. Treatment of EGPA requires immunosuppression mainly with glucocorticoids, but other agents, such as azathioprine, can be also considered.
Keywords:
Eosinophilia
Eosinophilic granulomatosis with polyangiitis
Eosinophilic myocarditis
Eosinophilic granulomatosis with polyangiitis (EGPA) is one of the various causes of hypereosinophilic syndromes. It is a rare multisystemic autoimmune disorder mainly affecting small-size and medium-size blood vessels by eosinophilic inflammation.1 Cardiac involvement – impaired LVEF, valvular insufficiencies, pericardial effusion – can represent one of its components in up to 50% of cases.2 The presence of myocarditis was described in up to 27% of EGPA patients2 and it is its most serious complication.
We present a case of a 36-year-old woman with a history of bronchial asthma and no previous cardiovascular disease. She had a history of polytopic allergies and had recently undergone a paranasal polypectomy in general anesthesia with a hypersensitive reaction consisting of severe prolonged bronchospasm. In the following month prior to index hospitalization, she had developed fatigue, productive cough, an upper and lower dyspeptic syndrome with lack of appetite, weight loss, and joint pain.
The patient presented at the emergency department with acute onset of chest pain upon inspiration and positional change. The initial examination revealed high sensitivity troponin I (hs-TnI) elevation, electrocardiogram changes (ST elevations in precordial leads and ST depressions in inferior and lateral leads), and mild systolic dysfunction (left ventricular ejection fraction [LVEF] 40%) with diffuse hypokinesis and asmall circular pericardial effusion on bedside echocardiography. There was leukocytosis with significant eosinophilia (26.2 × 109/l leukocytes with 11.170 × 109/l eosinophils) in the initial blood count, later laboratory examinations showed a progressive increase in hs-TnI with a maximum of 12 000 ng/l (normal range: 0.0–15.6 ng/l). Serial echocardiography revealed a progression of systolic dysfunction (LVEF 25–30%,right ventricle with normal function), clinically the patient developed signs of low cardiac output requiring transient administration of low-dose vasopressor and dopamine. Coronary angiography was performed with a normal finding. Cardiac magnetic resonance showed late gadolinium enhancement (LGE) non-homogeneously distributed in the interventricular septum in the subendocardium of both right and left ventricle and in the anterior and lateral wall of the left ventricle, where the LGE was in the subendocardium and also mid-myocardium (Fig. 1). There were also discrete changes in the wall of the right ventricle. The findings were consistent with a diagnosis of acute myocarditis.

A right ventricular endomyocardial biopsy was performed. The histological findings revealed acute eosinophilic myocarditis with a significant component of myocyte necrosis, without obvious signs of vasculitis or presence of granulomas (Fig. 2).
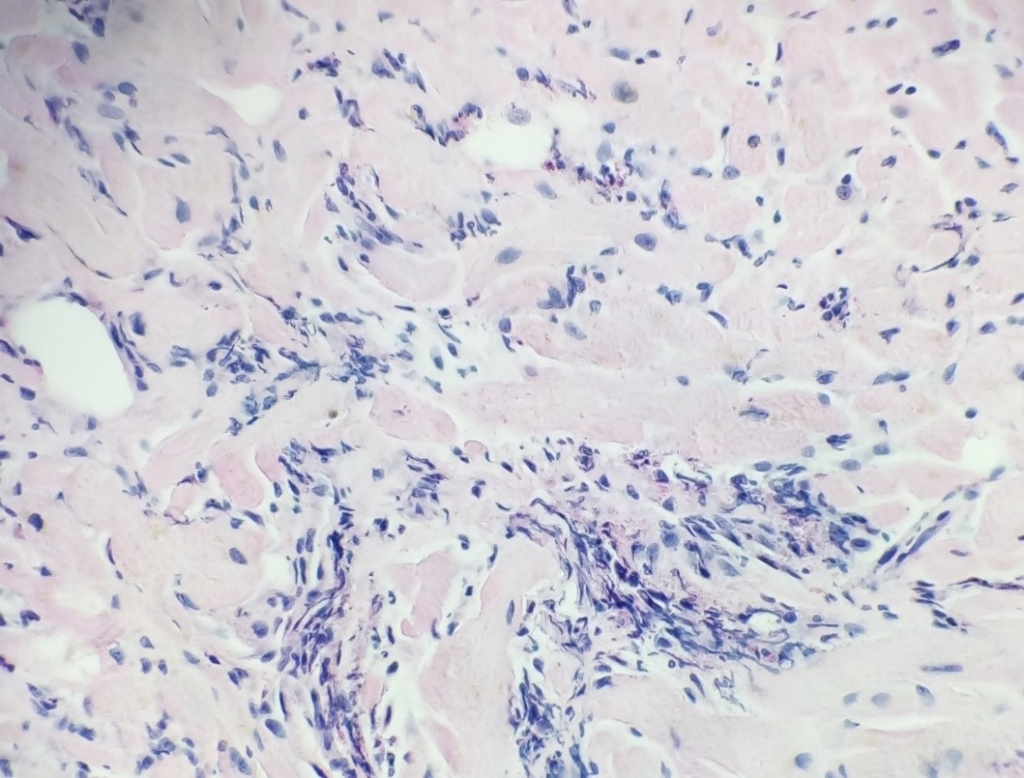
Several etiologies of the condition were considered. Firstly, we ruled out infectious agents: PCR from the acquired tissue excluded viral infection, PCR analysis of common respiratory viruses in nasopharyngeal smear, repeated hemocultivation, repeated stool parasitology analysis, rectal swab cultivation and serology of toxoplasmosis and aspergillosis were all negative. A sternal puncture was performed with the finding of isolated eosinophilia, without signs of atypical granulation, a genetic investigation focused on excluding primary hypereosinophilic syndromes yielded a negative result. The specialist concluded that a primary hematological pathology is unlikely. We also considered myocarditis as a part of a severe allergic reaction (hypersensitive myocarditis) to an unknownnoxa during general anesthesia in the patient’s premorbidity, which was not compliant with the histological findings.
A diagnosis of EGPA, formerly known as the Churg–Strauss syndrome) was discussed since the beginning of the hospitalization. We hesitated due to the complete absence of the most commonly described signs of this disease: chest X-Ray opacities, ANCA antibodies positivity and granulomas and vasculitis in histological samples. However, after completion of all examinations, we decided upon EGPA as the final diagnosis.
Intravenous corticosteroid therapy (methylprednisolone) was then commenced by an initial dose of 1 mg/kg per day of prednisone equivalent with subsequent regression of leukocytosis and eosinophilia, an improvement of left ventricular systolic function (LVEF 55%) and with overall clinical amelioration of the patient. Standard heart failure medical therapy was initiated after hemodynamic stability was achieved. The dose of corticosteroids was reduced and administered orally. Long-term heart failure medical therapy was limited by the patient’s tendency to hypotension, due to one non-sustained ventricular tachycardia registered during hospitalization a beta-blocker was preferred as a sole medication. The patient was discharged with a plan of gradual reduction of corticosteroid dose by 5 mg per week.
One month after hospital discharge the LVEF was still within a normal range and the patient had no clinical signs of heart failure or other complaints. Corticosteroid doses were reduced as planned and beta-blocker therapy was discontinued.
Six months after the hospitalization, while corticosteroid therapy doses were detracted, a worsening of LV systolic function occurred (LVEF 40%). The patient had no clinical signs of worsening of the heart failure and hs-TnI level was not elevated. A beta-blocker was reintroduced into medical therapy.
At nine-month follow-up the patient worsened clinically, with signs of asthma exacerbation, rhinitis, and elevation of eosinophils in the blood count. The LVEF remained unchanged. The dose of corticosteroids was increased with poor effect, therefore another immunosuppressive agent – azathioprine– was added to the medication. The initial dose was 100 mg daily and was subsequently increased to 150 mg daily due to insufficient effect. On the combination of a maintenance dose of corticosteroids (10–15 mg daily) and 150 mg of azathioprine, the patient is now clinically stable, LVEF remains the same.
The fluctuation of the patients’ clinical condition can be partly due to seasonal allergic rhinitis and asthma exacerbation during minor respiratory tract infections. However, the crucial role in the course of this chronic inflammatory disease plays the dosage of immunosuppressive therapy. Since long-term corticosteroid treatment burdens patients with serious side effects, there is a continuous attempt to retract the doses while still having good control over the disease.
The changes in the LVEF could be influenced by the quite early retraction of the already limited heart failure medication. Also continuous myocardial inflammation and fibrotization of the necrotic tissue probably took place simultaneously with the overall worsening of the disease. The published data concerning cardiac involvement in EGPA patients in clinical remission showed there are signs of ongoing myocardial inflammation in more than 25% of cases based on magnetic resonance imaging.2,3The limited data on long-term follow up of EGPA patients with cardiac involvement show predominantly favorable outcomes, however, patients with myocarditis seem to have more severe prognosis2,3.
This case report highlights the fact that eosinophilic myocarditis, can be the first recognized manifestation of EGPA.2The diagnosis of EGPA can be intricate since the clinical characteristics develop inconsistently and can also be consecutive. Vasculitis, the pathognomonic feature of EGPA is commonly not present until later phases and patients with cardiac involvement seem to be less likely to have the otherwise common ANCA antibodies2.
The most widely used set of criteria for EGPA diagnosis is The American College of Rheumatology criteria, which defines the probability of the diagnosis based on the presence of the following findings:4 bronchial asthma, eosinophilia (defined as >10% of eosinophils in blood count), neuropathy, migrating or transient opacities on chest X-Ray or CT scan, abnormalities of paranasal sinuses and histological proof of eosinophilecumulation in extravascular space. The presence of 4 out of 6 of above-listed findings (bronchial asthma, eosinophilia, paranasal polyps, and eosinophilic myocarditis) in our patient confirms the diagnosis of EGPA with 85% sensitivity and 99.7% specificity.
EGPA is one of the various causes of hypereosinophilic syndromes. It is a rare multisystemic autoimmune disorder and cardiac involvement can represent one of its components. The suspicion should be raised in patients with symptoms and signs of myocarditis and/or pericarditis or heart rhythm abnormalities with eosinophilia in blood count, with a history of asthma, allergic rhinitis, abnormalities in the paranasal sinuses and neuropathy.The diagnosis can be intricate since the clinical characteristics develop inconsistently and can also be consecutive. Vasculitis is commonly not present until later phases of the disease and patients with cardiac involvement seem to be less likely to have ANCA antibody positivity.2Anendomyocardial biopsy should be performed to prove the diagnosis of eosinophilic myocarditis in clinically relevant cases.5 Treatment of EGPA requires immunosuppression mainly with glucocorticoids, but other agents can be also considered.
Conflict of interest
The authors declare no conflict of interest.
Education: General medicine, MedicalSchoolof Charles University, Prague
Professional experience: 2015–now –Department of Cardiology, Motol University Hospital, Prague
2014–2015 – Department of Pediatrics, Motol University Hospital, Prague