




J. Naar, A. Krebsová, A. Krüger, D. Vondráková, M. Janotka, J. Šírek, P. Votýpka, M. Macek, P. Neužil, P. Ošťádal
Souhrn
Diagnostika příčiny oběhové zástavy u pacientů do 35 let nemusí být snadná. Řada onemocnění vedoucích k oběhové zástavě v nižším věku je hereditárních, s monogenním typem dědičnosti – kardiomyopatie, kanálopatie. Jejich odhalení znesnadňuje i nízká penetrance kauzální genové mutace a rozdílná expresivita. V případě arytmogenní kardiomyopatie je náhlá srdeční smrt či oběhová zástava často první manifestací onemocnění, před rozvinutím výraznějšího strukturálního postižení srdce. Kazuistické sdělení prezentuje případ 27letého sportovce, u kterého došlo k oběhové zástavě s úspěšnou kardiopulmonální resuscitací během soutěžního fotbalového utkání. Přestože na základě provedených zobrazovacích vyšetření nebylo patrné strukturální srdeční onemocnění a nebyla ani naplněna diagnostická kritéria arytmogenní kardiomyopatie, genetické vyšetření formou sekvenování DNA nové generace prokázalo mutaci genu pro desmosomální protein plakophilin 2. Mutační varianty tohoto proteinu patří k nejčastějšímu molekulárnímu podkladu arytmogenní kardiomyopatie. Rozšíření genetického testování v kardiologii může mít výrazný vliv nejen na zpřesnění diagnostiky příčiny oběhové zástavy u přeživších mladých nemocných, ale i na rizikovou stratifikaci blízkých příbuzných probanda.
© 2019, ČKS.
Klíčová slova:
Arytmogenní kardiomyopatie pravé komory
Katecholaminergní polymorfní komorová tachykardie
Náhlá srdeční smrt
Sekvenování DNA nové generace
Abstract
Diagnosing of the cause of out-of-hospital cardiac arrest in young patients ˂ 35 years old is challenging. Some disorders leading to cardiac arrest have monogenic type of heredity – cardiomyopathies, channelopathies. Diagnostic process may be complicated by low penetrance of causal gene mutation and variable expression. Sudden cardiac death or cardiac arrest often appear as the first clinical manifestation or arrhythmogenic cardiomyopathy before the development of obvious structural heart disease. The case re- port presents 27-year-old male athlete who underwent out-of-hospital cardiac arrest with successful cardiopulmonary resuscitation during soccer match. Despite the fact that there were no signs of structural heart impairment according to cardiac imaging and diagnostic criteria for arrhythmogenic right ventricular cardiomyopathy were not fulfilled, genetic testing using next-generation DnA sequencing revealed gene mutation in desmosome protein plakophilin 2, which represents the most frequent molecular background of arrhythmogenic cardiomyopathy. Extension of molecular testing in cardiology may contribute to more precise detection of the causal disease in young surviving patients who experienced cardiac arrest, as well as better risk stratification of their relatives.
Keywords:
Arrhythmogenic right ventricular cardiomyopathy
Catecholaminergic polymorphic ventricular tachycardia
Next-generation DNA sequencing
Sudden cardiac deat
Ischemická choroba srdeční je v rozvinutých zemích dominující příčinou náhlé srdeční smrti (NSS) a mimonemocniční oběhové zástavy (OHCA) u pacientů ve věku nad 35 let.
U mladších jedinců, včetně sportovců, patří mezi hlavní důvody OHCA dědičné kardiomyopatie, anomální odstupy koronárních tepen, specificky při sportu komoce srdce, dále pak zánětlivá onemocnění myokardu a kanálopatie.2,3 Mnohá z těchto onemocnění jsou hereditární, s mendelovským typem dědičnosti, převážně autosomálně dominantním. Kromě konvenčních kardiologických vyšetření, jako je 12svodová elektrokardiografie (EKG), echokardiografie či selektivní koronarografie (SKG), jsou dalšími diagnostickými nástroji pokročilejší zobrazovací metody srdce, zejména magnetická rezonance (MR), dále vyšetření věnčitých tepen výpočetní tomografií (CT koronarografie), dynamický zátěžový EKG test a signálově zprůměrované EKG (SAEKG). Selektivněji pak indikujeme invazivní elektrofyziologické vyšetření s použitím provokačních farmakologických testů a při podezření na infarkt myokardu vlivem paradoxní embolizace případně i jícnovou echokardiografii. Často však není kauzální onemocnění zřejmé ani po provedení této palety vyšetření. V poslední době může ke stanovení správné diagnózy při- spět genetické vyšetření pomocí sekvenování DNA nové generace.4
Sedmadvacetiletý, dosud zdravý, aktivně sportující muž, anamnesticky po krátké ztrátě vědomí v osmi letech věku při běhu, prodělal OHCA při soutěžním fotbalovém utkání. Bez prodlevy byla zahájena laická kardiopulmonální resuscitace (KPR), vykonávaná ovšem zdravotníkem – na stadionu přítomným lékařem. Po příjezdu posádky zdravotnické záchranné služby (přibližně po deseti minutách) bylo pokračováno v rozšířené KPR, dýchací cesty byly zajištěny orotracheální intubací. Prvním detekovaným rytmem byla fibrilace komor, která vykazovala refrakteritu k defibrilaci – celkem aplikováno deset externích výbojů. K obnově oběhu došlo po 20–25 minutách od zástavy. Pacient byl následně na umělé plicní ventilaci transportován na urgentní příjem oblastní nemocnice. Zde bylo provedeno vyšetření mozku výpočetní tomografií, které vyloučilo intrakraniální krvácení i jiné ložiskové změny. Laboratorně byla přítomna těžká metabolická acidóza (koncentrace laktátu v žilní krvi 19 mmol/l, pH krve 6,7), na EKG byl zachycen krátký běh polymorfní komorové tachykardie (obr. 1).

Pacient byl následně odeslán do našeho kardiocentra. Orientační vstupní transthorakální echokardiografické vyšetření neprokázalo jasné strukturální onemocnění srdce, nebyly přítomny známky akutního cor pulmonale ani jiná patologie vysvětlující OHCA. Interval QT na EKG byl v normě. Pro přechodné elevace úseků St ve svodech V – bylo přistoupeno bezodkladně k provedení SKG, kde jsme na věnčitých tepnách konstatovali normální nález, bez patrné odstupové anomálie. U pacienta byla zahájena řízená terapeutická hypotermie endovaskulárním zpětnovazebným systémem na cílovou teplotou 33 °C, později navýšenou na 34 °C pro výraznou bradykardii, s udržováním cílové teploty po dobu 24 hodin. Základní toxikologické vyšetření bylo negativní. Pacient po ukončení terapeutické hypotermie a vysazení analgosedace navázal kvalitní kontakt a bylo možné ho po dvou dnech od oběhové zástavy extubovat prakticky bez neurologického deficitu. Laboratorně byla zaznamenána výrazná elevace markerů myokardiálního poškození s vrcholovými hodnotami CK-MB mass (hmotnostní koncentrace MB izoenzymu kreatinkinázy) > 600 µg/l a srdečního troponinu I > 75 000 ng/l s následným rychlým poklesem. MR srdce neprokázala přesvědčivé známky pro myokarditidu, v pozdním snímání nebylo zachyceno sycení gadoliniovou kontrastní látkou neischemického ani ischemického typu. Rovněž nebyla splněna MR kritéria pro arytmogenní kardiomyopatii (AKMP) (video 1).

Vedlejším nálezem při vyšetření MR byly známky aspirační pneumonie, což podporovala i dynamika zánětlivých parametrů. Plicní infekce byla na základě kultivačního nálezu z tracheálního aspirátu přeléčena meropenemem po dobu osmi dní. Elektrofyziologické vyšetření neprokázalo komorovou preexcitaci, programovanou stimulací komor byly méně agresivním protokolem indukovány nesetrvalé polymorfní komorové tachykardie do deseti komplexů QRS, při agresivním protokolu pak rychlá setrvalá monomorfní komorová tachykardie tvaru blokády pravého Tawarova raménka (RBBB) (obr. 2).

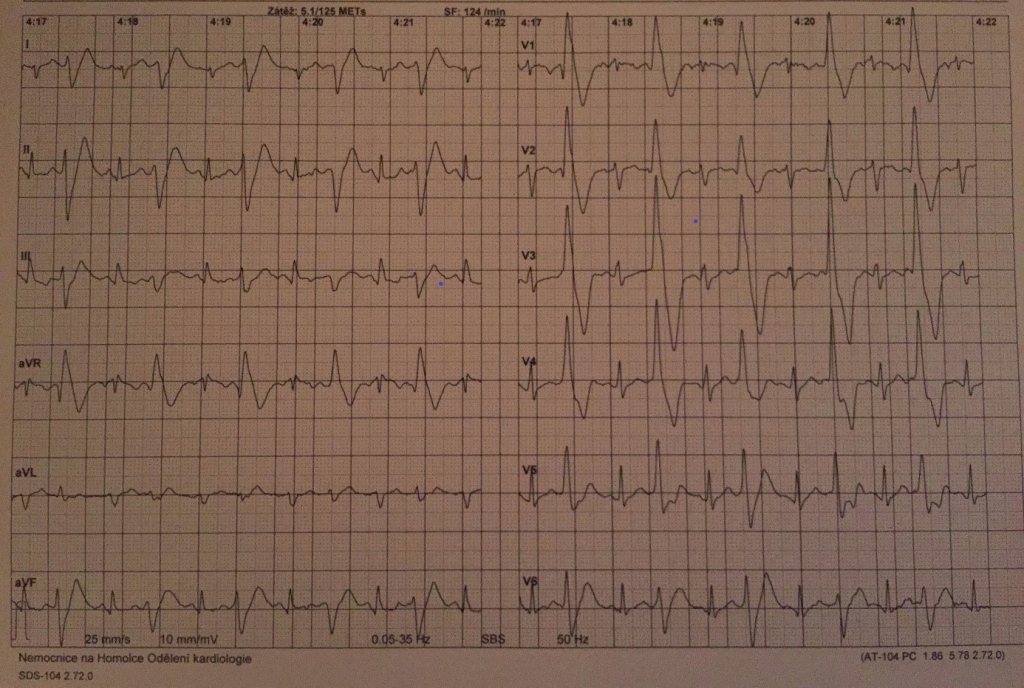
Ajmalinovým testem nebyly demaskovány EKG změny charakteristické pro syndrom bratří Brugadových. Nález na SAEKG byl hraniční, co se týče přítomnosti pozdních komorových potenciálů. Pacient byl propuštěn 14. den hospitalizace na terapii beta-blokátorem – metoprolol 25 mg/den s výhledem další uptitrace dávky, byl poučen o režimových opatřeních, zejména zanechání sportu na soutěžní úrovni. Den před propuštěním mu byl implantován kardioverter-defibrilátor v sekundární prevenci NSS. Ambulantně byl ještě doplněn zátěžový EKG test, při kterém došlo od stupně zátěže 150 W k opakovanému reproducibilnímu vyvolání bigeminie ne zcela monotopních komorových extrasystol tvaru RBBB, což bylo doprovázeno presynkopálním stavem (obr. 3).
Pacienta jsme odeslali do kardiogenetické ambulance, kde po klinicko-genetické konzultaci a získání informovaného souhlasu byla metodou sekvenování DNA nové generace (Illumina, roche) zjištěna velmi pravděpodobná až jistá příčinná varianta DNA v genu pro plakophilin 2 (PKP2, c. 1689-1G>c, třída 5). Gen PKP2 a změny v něm představují nejčastější molekulární příčinu AKMP. Následné kardiogenetické vyšetření matky pacienta odhalilo parciální fenotyp AKMP a potvrdilo nosičství identické varianty DNA v genu pro PKP2. naopak u bratrance z otcovy strany, který trpěl opakovanými palpitacemi, byla příčinná varianta DNA v genu pro PKP2 vyloučena. Podkladem jeho obtíží byla AV nodální reentry tachykardie.
Klinický případ popisuje mimonemocniční oběhovou zástavu při sportu u mladého pacienta bez zřejmého strukturálního onemocnění srdce dle echokardiografie a MR, u kterého až genetické vyšetření odhalilo příčinnou diagnózu – AKMP.
AKMP je geneticky podmíněné onemocnění, převážně s autosomálně dominantním typem dědičnosti, s rozdílným regionálním výskytem. Nejčastějším molekulárním podkladem je mutace genů kódujících proteiny buněčného spojení kardiomyocytů. V českých a evropských souborech jsou nejvíce zastoupeny mutace genu pro PKP2. Kauzální mutaci je u dědičné AKMP možno odhalit pomocí molekulárně genetického vyšetření až u 73 % pacientů.5 Téměř u čtvrtiny jedinců může být první klinickou manifestací AKMP NSS,6 typicky mezi 30. a 40. rokem věku. K náhlé srdeční smrti dojde nejčastěji při běžných denních aktivitách. Během fyzické zátěže je však riziko NSS přibližně pětkrát vyšší a k úmrtí při sportu dochází v nižším věku – mezi 20. a 30. rokem.7 Systematické kardiogenetické vyšetření příbuzných nemocného umožní identifikaci mutací postižených jedinců a jejich následnou kardiologickou dispenzarizaci formou kaskádového rodinného screeningu. Tímto opatřením je možné redukovat riziko NSS geneticky zatížených příbuzných probanda.8
Po provedení základních vyšetření padlo největší podezření na katecholaminergní polymorfní komorovou tachykardii (CPVt) jako příčinu OHCA. Pro tuto jednotku svědčila kromě absence strukturálního onemocnění srdce zejména oběhová zástava při zátěži, anamnéza námahové synkopy v dětství, fibrilace komor s obtížnou defibrilací jako vstupní rytmus při KPR, záchyt polymorfní komorové tachykardie po obnově oběhu, reprodukce komorových arytmií (bigeminicky vázaných extrasystol) při dynamickém zátěžovém testu. Mutace v genech spojených nejčastěji s CPVt (pro ryanodinový receptor a kalsekvestrin) však nebyla prokázána9 a naopak byla identifikována varianta v genu kódujícím protein desmosomu korespondující s onemocněním AKMP.
Stanovení diagnózy AKMP je komplikované. Vychází z diagnostických kritérií, k jejichž revizi došlo v roce 2010.10 Za zahrnují anamnestická data, histologický nález z endomyokardiální biopsie, elektrokardiografickou charakteristiku komorové depolarizace a repolarizace, záchyt komorových arytmií z oblasti pravé komory a morfologická kritéria získaná zobrazovacími vyšetřeními – echokardiografií a MR. Přestože jsme s touto jednotkou v diferenciálně diagnostickém procesu počítali a aktivně po diagnostických kritériích AKMP pátrali, k jejich naplnění zdaleka nedošlo. Klidové 12svodové EKG nebylo zcela normální – obraz inkompletního RBBB s elektrickou osou srdeční ve frontální rovině doprava, bez typické vlny epsilon, s určitými repolarizačními změnami, nicméně pozitivními vlnami T ve svodech V2 a V3. Záznam pozdních potenciálů pomocí SAEKG byl vyhodnocen jako hraniční – trvání terminálního nízkoamplitudového signálu 38 ms, střední kvadratická voltáž terminálních 40 ms 21 µV. Vyšetření MR nenaplnilo kritéria pro diagnózu AKMP. Bylo sice splněno MR kritérium pro end-diastolický objem pravé komory, ale nebyla přítomna druhá podmínka, tedy regionální akineze, dyskineze či dyssynchronie kontrakce pravé komory. Komorová tachykardie indukovaná programovanou stimulací komor i komorové extrasystoly vyvolané dynamickou zátěží měly navíc tvar blokády pravého Tawarova raménka, nikoliv levého. Z výše uvedeného není vyloučeno, že pacient má oboustrannou formu AKMP.
Zákeřnost AKMP spočívá v tom, že arytmická manifestace může předcházet rozvoji zjevného strukturálního postižení srdce, a ani cílený kardiologický screening sportovců nemusí mít dostatečnou senzitivitu k odhalení onemocnění a NSS může být jeho prvním projevem. Další faktor, který znesnadňuje detekci tohoto monogenně dědičného onemocnění, je nízká penetrance5 a variabilní expresivita kauzální mutace. Toto potvrzuje i naše kazuistika. U pacienta nebyla přítomna anamnéza náhlé srdeční smrti či synkopy u blízkých příbuzných, přesto genetické testování prokázalo asymptomatické nosičství genové mutace u matky s částečně vyjádřeným fenotypem AKMP. Při jejím základním kardiologickém vyšetření byly shledány diagnostické repolarizační změny na 12svodovém EKG (obr. 4).

Hodnoty matčina SAEKG malé diagnostické kritérium nesplňovaly (obr. 5), echokardiografie prokázala dilataci výtokového traktu pravé komory.

Dřívější stanovení diagnózy AKMP u tohoto pacienta by zřejmě neovlivnilo léčebný postup, ale znalost kauzální genové mutace má zásadní vliv na prevenci NSS u jeho příbuzných.
Rovněž příčina náhlé srdeční smrti mladých, dosud zdravých jedinců zůstává i po provedené soudní pitvě u nezanedbatelné části nevyjasněna.11 Pokud sekce příčinu jednoznačně neozřejmí a existuje podezření na konkrétní kardiomyopatii nebo kanálopatii, je opodstatněné cílené post-mortem genetické vyšetření (doporučení ve třídě IIa dle nedávných odborných klinických guidelines Evropské kardiologické společnosti).12
Větší uplatnění genetického vyšetření v kardiologii by mohla přinést iniciativa centralizace pacientů po OHCA – vznik center péče o nemocné po srdeční zástavě. V popsaném případu nebylo odeslání pacienta zcela v souladu s touto koncepcí vyjádřenou společným stanoviskem několika českých odborných společností.13 Podle ní by měla posádka zdravotnické záchranné služby u pacienta po OHCA bez zjevné nekardiální příčiny kontaktovat specializované centrum, do kterého má být nemocný transportován přímo z místa zásahu.
Aktuálnost tématu molekulárně genetického vyšetření v kardiologii jen podtrhuje vznik samostatné pracovní skupiny Kardiogenetika v rámci České kardiologické společnosti.
Prezentované kazuistické sdělení ukazuje, že vyhodnocení příčiny oběhové zástavy u přeživších mladých nemocných bez známého srdečního onemocnění a bez jasného strukturálního postižení srdce dle vstupních zobrazovacích vyšetření může být obtížné a standardními diagnostickými postupy nemusíme základní onemocnění odhalit, popřípadě nás navede k nesprávnému závěru. Molekulárně genetické vyšetření s využitím sekvenování DNA nové generace je účinný nástroj, který má potenciál diagnostický proces pozitivně ovlivnit. Neměli bychom ho však aplikovat paušálně, ale pouze při podezření na určitou dědičnou kardiomyopatii nebo kanálopatii. Jelikož výsledek genetického vyšetření může být negativní nebo ne- konkluzivní, je k jeho správné interpretaci nezbytné maximum informací ze systematicky a pečlivě provedených kardiologických vyšetření, včetně MR srdce, která dokáže příčinu OHCA ozřejmit v 19–33 % případů, a SAEKG a dynamického zátěžového testu, jež jsou u této skupiny pacientů nedostatečně využívány v diagnostickém procesu.14,15 Současně je velmi důležitý kaskádový rodinný screening, a to nejen pro zajištění primární prevence NSS u přímých příbuzných v riziku, ale i k validaci výstupů molekulárně genetického vyšetření.
Prohlášení autorů o možném střetu zájmů
Autoři prohlašují, že nemají střet zájmu.
Financování
Genetické vyšetření bylo finančně podpořeno z programového projektu MZ ČR s registračním číslem NV18-02-00237 a interního grantu IKEM G9039. Práce byla podpořena vnitřním grantem institucionální podpory MZ (NNH, 00023884), IG 180502.
Literatura
1. M. Hayashi, W. Shimizu, C.M. Albert, The spectrum of epidemiology underlying sudden cardiac death, Circulation Research 116 (2015) 1887–1906.
2. B. J. Maron, J. Shirani, L.C. Poliac, et al., Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles, Journal of the American Medical Association 276 (1996) 199–204.
3. N. Chandra, R. Bastiaenen, M. Papadakis, S. Sharma, Sudden cardiac death in young athletes: practical challenges and diagnostic dilemmas, Journal of the American College of Cardiology 61 (2013) 1027–1040.
4. M.J. Ackerman, S.G. Priori, S. Willems, et al., HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies, Europace 13 (2011) 1077–1109.
5. A. Krebsová, L. Piherová, J. Paděrová, et al., Dosavadní výsledky genetického vyšetření pacientů s ARVC/D v IKEM, pilotní studie, XXIV výroční sjezd ČKS 2016.
6. D. Dalal, K. Nasir, C. Bomma, et al. Arrhythmogenic right ventricular dysplasia: a United States experience, Circulation 112 (2005) 3823–3832.
7. A. Tabib, R. Loire, L. Chalabreysse, et al., Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia, Circulation 108 (2003) 3000–3005.
8. P. Charron, M. Arad, E. Arbustini, et al., Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases, European Heart Journal 31 (2010) 2715–2726.
9. A.A.M. Wilde, A. Amin, Channelopathies, genetic testing and risk stratification, International Journal of Cardiology 237 (2017) 53–55.
10. F.I. Marcus, W.J. McKenna, D. Sherrill, et al., Diagnosis of arrhythomogenic right ventricular cardiomyopathy/dysplasia. Proposed modification of the task force criteria, European Heart Journal 31 (2010) 806–814.
11. A. Mazzanti, S. O'Rourke, K. Ng, et al., The usual suspects in sudden cardiac death of the young: a focus on inherited arrhythmogenic diseases, Expert Review of Cardiovascular Therapy 12 (2014) 499–519.
12. S.G. Priori, C. Blomström-Lundqvist, A. Mazzanti, et al., 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), European Heart Journal 36 (2015) 2793–2867.
13. P. Ošťádal, R. Rokyta, M. Balík, et al., Centra péče o nemocné po srdeční zástavě. Společné stanovisko odborných společností: České asociace akutní kardiologie České kardiologické společnosti, České resuscitační rady, České společnosti intenzivní medicíny ČLS JEP, České společnosti anesteziologie, resuscitace a intenzivní medicíny ČLS JEP, Společnosti urgentní medicíny a medicíny katastrof ČLS JEP, Cor et Vasa 59 (2017) e196–e199.
14. C.C. Cheung, A.D. Krahn, The importance of a comprehensive evaluation of survivors of cardiac arrest, European Heart Journal 39 (2018) 1988–1991.
15. A.D. Krahn, J.S. Healey, V. Chauhan, et al., Systematic assessment of patients with unexplained cardiac arrest: Cardiac Arrest Survivors With Preserved Ejection Fraction Registry (CASPER), Circulation 120 (2009) 278–285.
V roce 2008 MUDr. Jan Naar absolvoval obor všeobecné lékařství na Lékařské fakultě v Plzni Univerzity Karlovy. Do roku 2010 pracoval na I. interní klinice Fakultní nemocnice Plzeň, od roku 2010 na Kardiologickém oddělení Nemocnice Na Homolce. V roce 2017 složil atestaci z kardiologie. Klinickou oblastí zájmu je akutní kardiologie, srdeční selhání, echokardiografie, výzkumnou oblastí zájmu nefarmakologická léčba srdečního selhání.



