




K. Rücklová, E. Honsová, P. Tomášek
Úvod: Aneurysma a disekce hrudní aorty představují vzácnou, ale důležitou příčinu úmrtí v mladém věku. Tato kazuistika popisuje případ mladého muže, který zemřel na tamponádu srdce při ruptuře hrudní aorty. Pitva zároveň odhalila mnohočetná aneurysmata středně velkých tepen. V kazuistice jsou diskutovány možné příčiny těchto mnohočetných cévních anomálií, především dědičná onemocnění pojiva. V případě suspektní dědičné příčiny je v rámci vyšetření post mortem indikována molekulární pitva a kardiogenetické poradenství v rodině.
Popis případu: Devětadvacetiletý muž zemřel náhle během letu na rupturu hrudní aorty. Pitva kromě srdeční tamponády odhalila i mnohočetná aneurysmata bazilárních a koronárních tepen s trombotickým uzávěrem ramus interventricularis anterior. Kromě geneticky podmíněné vaskulopatie mohou být příčiny podobných nálezů různorodé – od aterosklerózy a arteriální hypertenze až po různé typy vaskulitid. Popisujeme klinické a patologické charakteristiky jednotlivých onemocnění, aby se dostaly do povědomí kliniků i patologů.
Závěr: Ruptura hrudní aorty s asociovanými aneurysmaty nebo bez asociovaných aneurysmat středně velkých tepen u mladé osoby zasluhuje zvláštní pozornost, protože může být způsobena potenciálně dědičnou vlohou. Ta se navíc dědí autosomálně dominantním způsobem a může znamenat zvýšené riziko náhlé smrti u prvostupňových příbuzných. V našem případě jsme genetickou příčinu potvrdit nemohli, protože nebyla k dispozici vhodná tkáň pro analýzu DNA. Právě proto tato kazuistika podtrhuje, jak je důležité na dědičná onemocnění pojiva pomýšlet a uchovat vhodnou tkáň pro molekulární pitvu.
© ČKS, 2022.
Klíčová slova:
Aneurysma a disekce hrudní aorty
Koronární a intrakraniální aneurysmata
Příčiny
Background: Thoracic aortic disease is a rare but important cause of death that may affect young individuals. We present a case of a young man who died of thoracic aortic rupture associated with multiple aneurysms of medium-sized arteries. Possible causes of this multivascular pathology are discussed here with emphasis on potential genetic etiology and its implications for post mortem investigations including molecular autopsy and cardiogenetic counselling in the family.
Case presentation: A 29-year-old man died suddenly due to thoracic aortic rupture during an air flight. His autopsy, in addition, revealed multiple aneurysms in the basilar and coronary arteries with a thrombotic occlusion of an aneurysm in the left anterior descending artery. Apart from genetic etiology the causes of similar findings range from atherosclerosis and hypertension to various inflammatory diseases. The clinical and pathological features of relevant diseases are outlined here to raise awareness of them among clinicians as well as pathologists.
Conclusions: The finding of thoracic aortic rupture in a young person with or without associated medium-sized artery aneurysms deserves special attention due to its potentially heritable etiology with an increased risk of sudden death for first degree relatives. In our case we could not confirm genetic etiology due to lack of suitable material for DNA analysis. We therefore emphasize the importance to consider heritable connective tissue disorders in similar cases and to retain suitable tissue for molecular autopsy.
Keywords:
Causes
Coronary aneurysms
Intracranial aneurysms
Thoracic aortic disease
Address: MUDr. Kristina Rücklová, Ph.D., Department of Pediatrics, University Hospital Královské Vinohrady, Šrobárova 1150/50, 100 34 Prague 10, e-mail: kristina.rucklova@fnkv.cz
Please cite this article as: Rücklová K, Honsová E, Tomášek P. Thoracic aortic rupture associated with multiple aneurysms of medium-sized arteries: case report. Cor et Vasa Case Reports
2022;5:7–11.
Thoracic aortic disease (TAD) with rupture is an important cause of sudden death accounting for approximately 7% of sudden cardiac death cases among people aged 1–35 years.1 Major risk factors for TAD include ageing, increased mechanical forces acting on aorta such as poorly controlled hypertension or stimulant abuse and an underlying genetic predisposition that disrupts aorta’s elastic properties rendering it more susceptible to dilation and dissection.2 Especially in young persons, a heritable predisposition is predominantly responsible for TAD.3 Other possible causes of thoracic aortic aneurysm and rupture in young individuals include infectious or non-infectious inflammation, with Takayasu arteritis being the most common vasculitis that affects aorta in patients below 50 years of age.4
Patients with heritable or inflammatory TAD may develop aneurysms elsewhere in the arterial tree including coronary and cerebral arteries. However, except for reports on Loeys–Dietz and the vascular type of Ehlers–Danlos syndrome there is a limited number of reports addressing this association.5–7
The patient presented here was recruited from a retrospectively collected cohort of sudden cardiac death cases aged 1–40 years autopsied in the Czech Republic. He was the youngest TAD case in our cohort and stands out due to the unusual autopsy finding of TAD associated with multiple aneurysms in the basilar and coronary arteries. The exact etiology of these morphological changes could not be determined as no data regarding his previous medical history were available and genetic testing could not be performed due to lack of suitable tissue. Nevertheless, this case report provides us with an opportunity to discuss the differential diagnosis of such findings and consider practical implications for post mortem investigations in similar cases.
A 29-year-old healthy looking man of Caucasian ethnicity collapsed suddenly during an airplane flight in 2015. Cardiopulmonary resuscitation provided by the airplane personnel was unsuccessful. His ECG showed asystole with no reaction to repeated adrenaline administration.
The young man was 186 cm (+0.8 SD above average) tall and obese (BMI 37). His fingers were remarkably long and thin. He had normal facial features, no corneal opacities, no palatal cleft, bifid uvula or chest deformity, nor did he have any other skeletal abnormalities. Apart from two hypopigmented scars on the left shoulder and arm, his skin was unremarkable. It was not particularly thin or translucent and no striae were noticeable. Except for resuscitation induced rib fractures with right lung hematoma and brain and lung edema, no pathological organ involvement was noted. Increased fragility of intestinal wall was not observed and the patient had no liver or kidney cysts.
Thoracic aortic rupture within an aneurysm with cardiac tamponade was revealed as the cause of death. The aneurysm was saccular 3 × 1.5 × 1.5 cm in size and located above the aortic valve. The aortic wall rupture extended 4 cm in length and led to cardiac tamponade. The pericardial sac contained 750 ml of blood. The aorta contained only minor fibrous plaques on macroscopic examination. The aortic valve was tricuspid.
Additional pathological findings included multiple fusiform basilar artery aneurysms along the whole length of the vessel with one aneurysm 1 × 0.5 cm in diameter. Cerebral arteries showed no significant atherosclerosis.
Another fusiform aneurysm was revealed in the left anterior descending (LAD) artery near the branching of the left main coronary artery, where the vessel circumference reached 20 mm and the aneurysm was completely occluded with an old organized thrombus of 3.5 × 2 × 1.3 cm. A pale post-ischemic intramural scar of 2 × 1 cm was noted in the interventricular septum. The scar signified a past myocardial infarction that could have been related to the thrombus in LAD. Circumflex coronary artery was also locally dilated. Minor atherosclerotic plaques were present in the coronary arteries that caused only insignificant stenoses of their lumen.
Abdominal aorta and other arteries did not show aneurysmal dilations and/or atherosclerotic changes.
The heart was concentrically hypertrophic. Its weight of 680 g exceeded the predicted BSA related normal heart weight of 455.3 g (range 370.1–540.6 g)8 and the left ventricular wall thickness was 16 mm also slightly exceeding two standard deviations above the mean of 12.6 mm.9 Myocardial disarray pathognomonic for hypertrophic cardiomyopathy was not present.
The patient was tested negative for alcohol.
Aortic and coronary artery specimens were stained with hematoxylin-eosin (HE) and Van Gieson (VG) with modification for additional elastin detection.
Hemorrhage and smooth muscle disorganization was observed in the specimens of the aortic wall as well as elastic fiber fragmentation, focal elastic fiber loss and focal mucoid extracellular matrix accumulation10 (Fig. 1A and Fig. 1B). There were no atherosclerotic changes and no intimal calcifications in the aortic wall.
The specimen of the left anterior descending coronary artery showed an aneurysm with an organized thrombus. Neither aorta nor coronary arteries contained inflammatory infiltrates, granulomas or giant cells that would indicate inflammatory etiology.11,12
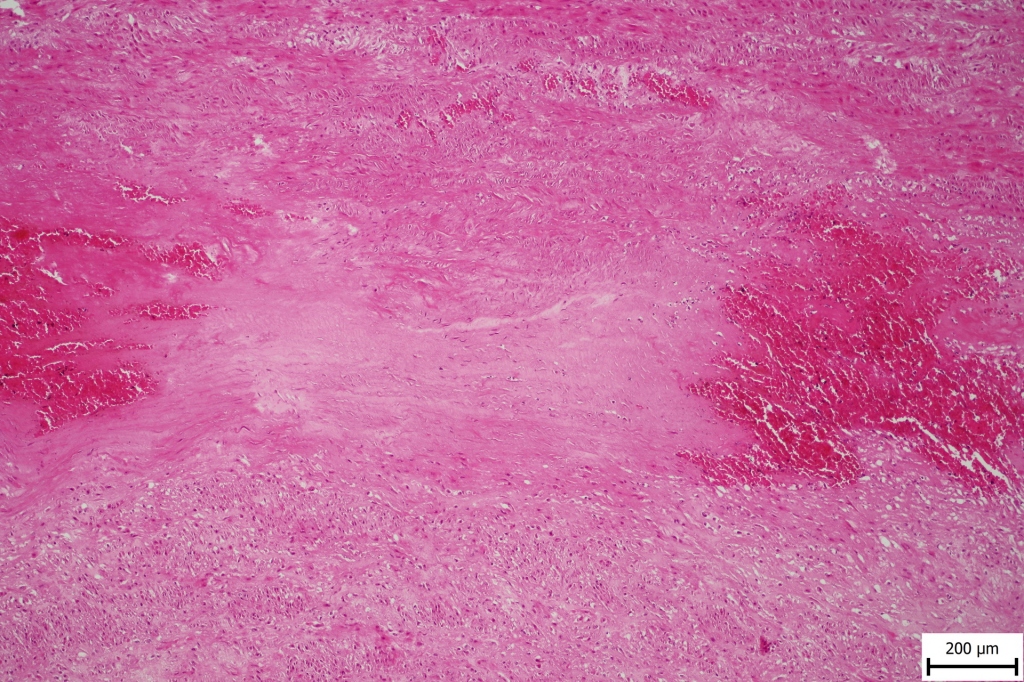
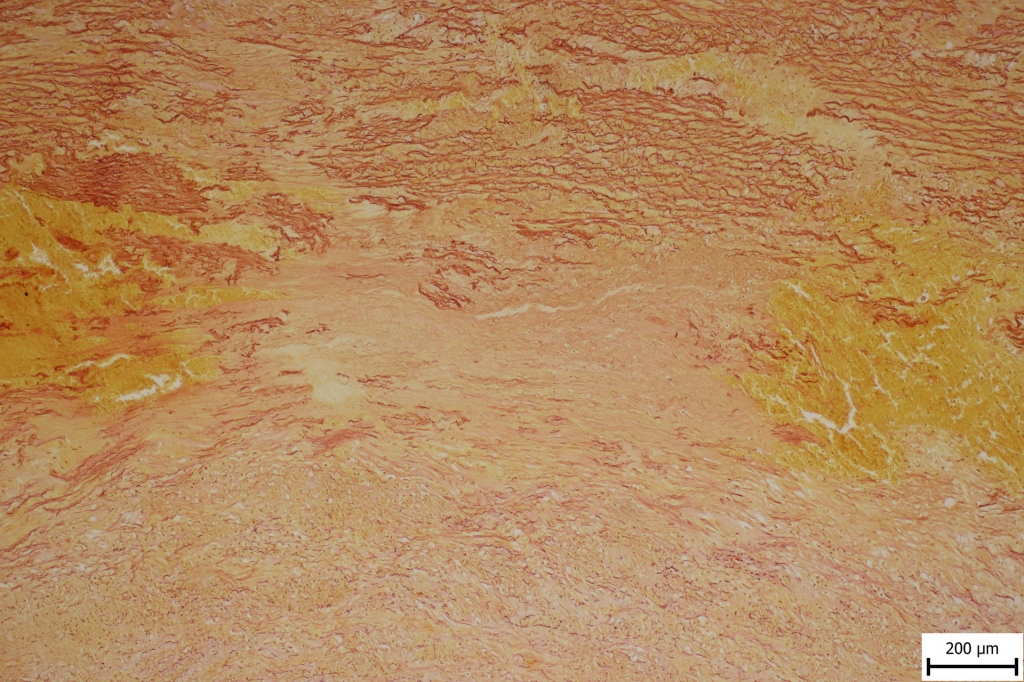
The patient reported here had a cardiac hypertrophy, which may suggest arterial hypertension as one of the major risk factors for thoracic aortic rupture. However, his young age, normal kidneys with smooth surface and additional vascular pathology with multiple basilar and coronary aneurysms make hypertension as a sole cause of these findings rather unlikely.
Atherosclerosis is the most common cause of coronary aneurysms13 and may be associated with aortic aneurysm and rupture. However, in contrast to abdominal aortic aneurysm, thoracic aortic aneurysm due to atherosclerosis is very rare.14 In addition, our patient was young and almost no atherosclerotic changes were observed in his aorta.
The young age of the patient suggests rather a genetic or inflammatory etiology of the vascular damage. Considering inflammatory diseases, the most likely culprit could be Kawasaki disease, the second most common cause of coronary aneurysms in adults. However, it has only rarely been associated with thoracic aortic aneurysms15,16 and thoracic aortic rupture has never been reported. Aneurysms in other medium-sized systemic arteries occur in about 2% of Kawasaki disease patients17 and their most frequent locations are brachial, axillary and iliac arteries.18 On the other hand, intracranial aneurysms have been observed only exceptionally. The inflammatory process actually seems to spare intracranial arteries.12 In our patient, the histological findings together with these facts make the diagnosis of Kawasaki disease very unlikely.
Another vasculitis, which typically affects aorta as well as coronary arteries, is Takayasu arteritis. It may manifest with thoracic aortic rupture and coronary arteries are involved in as many as 53% of these patients. Coronary aneurysms are present in 8% and coronary stenoses in the rest.19 Intracranial aneurysms including basilar aneurysms have been described in Takayasu arteritis as well.20 However, Takayasu arteritis typically affects young women of Asian ethnicity. The initial stage of the disease is clinically characterized by symptoms of systemic inflammation or acute aortic insufficiency. Transmural granulomatous inflammation with varying number of giant cells is typically found in histopathological specimens at this stage. The chronic phase is characterized by intimal fibrosis leading to vascular obstruction.4,11 However, the histopathological findings in our patient exclude the diagnosis of Takayasu arteritis.
Vasculitis involving large as well as medium-sized arteries may also occur in IgG4-related disease.11,21 However, this disease is usually manifested by tumor-like lympho-plasmocytic infiltrations in various tissues especially pancreas, liver, kidneys, salivary gland or orbital region, which was not observed in our patient. It mostly affects persons more than 50 years old,22 although much younger patients have also been reported.23 Autopsy findings and histopathological evaluation rule out this condition in our patient.
Similarly, we can also exclude ANCA-associated vasculitides which may though exceptionally involve large vessels including aorta.24
By exclusion and with respect to the young age of our patient we speculate that the most likely underlying etiology of the aortic rupture and multiple medium-sized artery aneurysms might be a pathogenic variant in one of the genes associated with heritable TAD (HTAD). Approximately 10% of patients with HTAD have a concomitant intracranial aneurysm25 and coronary aneurysms have also been reported5–7 supporting the possibility of a genetic cause in our patient.
Depending on the presence or absence of involvement of other organ systems, HTAD may be subdivided into syndromic and isolated HTAD. So far 11 genes, whose pathogenic variants confer a very high risk of TAD, have been identified. They encode proteins involved in maintaining elasticity and strength of the medial layer of the aorta, which is composed mainly of smooth muscle cells and elastin. On the cellular level, these proteins are mainly implicated in smooth muscle cell contraction and adhesion to the extracellular matrix (e.g. FBN1, COL3A1, ACTA2, MYH11, MYLK, PRKG1, MFAP5), transforming growth factor-β signaling pathway (e.g. TGFBR1/2, SMAD3/4) or smooth muscle cell metabolism.2 Pathogenic variants in these genes may be found in syndromic connective tissue disorders associated with TAD such as Marfan, Loeys–Dietz or vascular type of Ehler–Danlos syndrome as well as in isolated HTAD cases.
Our patient exhibited only arachnodactyly as a characteristic feature of syndromic form of HTAD. He had no other typical musculoskeletal or ocular abnormalities nor did he exhibit less common features such as liver or kidney cysts associated with Marfan syndrome.26 His habitus was not asthenic. This latter feature had historically been considered typical for Marfan syndrome but more recently it has been documented that as many as 26% of Marfan syndrome patients are obese.27 Hence, Marfan syndrome cannot be excluded in our patient as it is the most common syndromic connective tissue disorder associated with TAD. The other above mentioned syndromes may be associated with aneurysms throughout the arterial tree but their characteristic features such as bifid uvula, hypertelorism, translucent and hyperelastic skin, atrophic scars, chest deformities, short or tall stature, joint hypermobility or contractures3 were absent in our patient.
As no pronounced extravascular anomalies were observed, we consider non-syndromic HTAD to be the most likely diagnosis in our patient. We may hypothesize about a possible role of certain genes as some have been associated with TAD and additional vascular complications. For example, pathogenic variants in TGFBR2 gene predispose to TAD as well as intracranial and other arterial aneurysms. Another highly suspicious gene could be ACTA2 as its pathogenic variants lead to TAD together with early-onset coronary artery disease as seen in our patient who had a thrombotic occlusion of the left anterior descending artery.2 Pathogenic variants in ACTA2 gene are responsible for 14% of non-syndromic HTAD, thereby being the most frequently identified molecular mechanism in this subgroup.28
The greatest limitation of this case report is the fact that genetic testing could not be performed due to lack of tissue for DNA analysis. All paraphin blocks with myocardial samples were used for preparation of histological specimens. Another limitation is absence of medical records.
TAD in a young person should always be suspected of genetic etiology. In fact, approximately 20% of TAD are familial with an autosomal dominant pattern of inheritance thus conferring a 50% risk for first degree relatives.3 Further deaths in the families may be prevented if individuals at risk are identified and managed in time. Therefore, tissue from the deceased person should be retained for DNA analysis and the relatives should receive an adequate cardio-genetic counselling including a cascade genetic screening.29,30 In order to implement this approach into routine practice, guidelines for post mortem investigations of sudden cardiac death cases were approved by the Czech Forensic Medicine Society in 2020.
This case report provides forensic pathologists as well as clinicians with an overview of differential diagnosis of multi-vascular aneurysms, which may help them select patients for targeted genetic testing. The report also contributes to increased awareness of heritable cardiovascular diseases, which may prompt general practitioners to indicate cardio-genetic care in families with sudden cardiac death in the young. Lastly, the case presented here demonstrates that TAD may be associated with aneurysms elsewhere in the arterial tree especially in coronary arteries.31 This fact is not broadly recognized and coronary arteries are not routinely checked during follow up of young patients with TAD.
Acknowledgements
The authors are particularly grateful to the secretary of the Institute of Forensic Medicine at University Hospital Bulovka who provided them with an access to de-identified autopsy protocols of potential sudden cardiac death victims.
Conflicts of interest
None.
Funding
Supported by the Ministry of Health of the Czech Republic, grant nr. NV18-02-00237. All rights reserved.
Ethical statement
Ethics committee’s approval was not required as all patient’s data were strictly de-identified. The study did not involve living subjects nor did it involve any genetic analyses.
Authors’ contributions
All authors contributed to the design of the article. Kristina Rücklová was responsible for writing the manuscript. Eva Honsová assessed the histological samples and suggested the differential diagnosis of the histological findings. Petr Tomášek evaluated the autopsy protocol. Both co-authors commented on the first version of the manuscript and all authors approved the final version.
References
MUDr. Kristina Rücklová, Ph.D. Education and qualification: qualification in clinical pediatrics (2nd degree) – Nov 2010; qualification in pediatric cardiology – Jun 2009; qualification in clinical pediatrics (1st degree) – Jun 2005; Ph.D. studies in physiology and pathophysiology, Ph.D. degree, 3rd Faculty of Medicine, Charles University, topic: “Retrospective diagnosis of unrecognized causes of sudden death in infancy” – 2002–2008; 1st Faculty of Medicine, Charles University, Prague, M.D. degree – 1995–2002; Faculty of Medicine, Ludwig Maximilians University, Munich, Germany – 1999–2000; Faculty of Philosophy – English Philology, Charles University, Prague – 1995–1999
Clinical experience: pediatric cardiologist, part-time, Dept. of Pediatrics and Inherited Metabolic Disorders, General University Hospital, Prague – 2018 – now; chief physician at the unit for children up to 3 years of age and pediatric cardiologist, University Hospital Královské Vinohrady, Prague – 2014 – now; pediatric cardiologist, Children’s Heart Centre, Faculty Hospital Motol, Prague – 2011–2014; pediatrician and pediatric cardiologist, Dept. of Pediatrics, Faculty Hospital Motol, Prague – 2008–2011; resident in pediatrics, Dept. of Pediatrics, Faculty Hospital Královské Vinohrady, Prague – 2002–2007



